
Gerelateerde artikels
Syndroom van Gilbert: chronische, veelvoorkomende maar goedaardige leverziekte
In dit artikel
Syndroom van Gilbert: chronische, veelvoorkomende maar goedaardige leverziekte
- Syndroom van Gilbert: een veelvoorkomende maar goedaardige leverziekte
- Wat is het syndroom van Gilbert?
- Symptomen en gevolgen van het syndroom van Gilbert
- Behandeling van het syndroom van Gilbert
- Syndroom van Crigler Najjar
- Symptomen en gevolgen van het syndroom van Crigler Najjar
- Behandeling van het syndroom van Crigler Najjar
dossier
Het syndroom van Gilbert of ongeconjugeerde hyperbilirubinemie, is een chronische maar goedaardige leverziekte. Bij pasgeborenen is het een belangrijke oorzaak van geelzucht (gele huid, geelverkleuring van het oogwit).
Ongeveer 1 op de 20 mensen heeft dit syndroom. Maar slechts 1 op de 3 mensen met deze aandoening heeft ook klachten.
Het vergelijkbare maar veel zeldzamer Syndroom van Crigler Najjar kan daarentegen wél gevaarlijk zijn.
Wat is het syndroom van Gilbert?
Het syndroom van Gilbert (of ongeconjugeerde hyperbilirubinemie) is een erfelijke aandoening waarbij de lever onvoldoende bilirubine afbreekt. Bilirubine is een afvalstof die ontstaat bij de afbraak van rode bloedcellen.
Rode bloedcellen hebben een gemiddelde levensduur van ca. 6 weken. Nadien worden ze afgebroken en uit het lichaam verwijderd. Bij die afbraak wordt de rode kleurstof in het bloed, het hemoglobine, afgebroken tot bilirubine, een gele kleurstof die het lichaam verlaat via de urine en de ontlasting.
Om dat bilirubine te kunnen uitscheiden moet het in de lever wateroplosbaar worden gemaakt. Dat proces heet conjugeren. Daarvoor heeft de lever een bepaald eiwit (het UGT1Al enzym) nodig. Wanneer dat enzyme in onvoldoende mate aanwezig is of slecht werkt, kan het bilirubine niet ‘geconjugeerd’ worden en kan het niet uitgescheiden worden maar blijft het aanwezig in het bloed. Vandaar ook de naam ongeconjugeerde hyperbilirubinemie: een teveel aan niet-geconjugeerd bilirubine in het bloed. Dat leidt dan tot geelzucht met als typische kenmerken een gele huid en geelkleuring van het oogwit.
Erfelijke ziekte
Het syndroom van Gilbert is een erfelijke aandoening die autosomaal dominant of recessief wordt overgedragen.
• Autosomaal betekent dat het een niet-geslachtsgebonden aandoening is: mannen en vrouwen kunnen het syndroom doorgeven en jongens en meisjes hebben evenveel kans om ze van hun ouders te erven. Dit komt omdat het afwijkend gen niet op het chromosoom ligt dat het geslacht van het kind bepaald.
• Dominant betekent dat een erfelijke eigenschap kan overgeërfd worden zodra een van de ouders het defecte gen heeft, ook als de andere ouder wel het normale gen heeft.
Kinderen van een vader of moeder met een autosomaal dominant erfelijke aandoening krijgen via de eicel (de moeder) of via de zaadcel (de vader) van de aangedane ouder een van de twee genen waarop de aandoening kan zitten, Ze kunnen dus het normale gen of het afwijkende gen krijgen. Elke zoon of dochter heeft daarmee een kans van 50% om de erfelijke aandoening te erven.
Het kan ook gebeuren dat iemand een autosomaal dominant erfelijke ziekte heeft, terwijl beide ouders die ziekte zelf niet hebben. Dan is er meestal sprake geweest van een nieuwe verandering bij het kind zelf, een nieuwe mutatie.
• Recessief betekent dat een kind alleen met het syndroom van Gilbert kan geboren worden als beide ouders er zelf aan lijden of drager zijn. Dragers hebben zelf de ziekte niet en dus ook geen klachten.
Als beide ouders dragers zijn, is kans dat ook het kind de ziekte krijgt 25% en 50% dat het drager wordt (zonder de ziekte te hebben). De kans dat het kind de ziekte niet erft is eveneens 25%.
Lees ook: Erfelijkheid: Blauwe of bruine ogen?
Symptomen en gevolgen van het syndroom van Gilbert
De meeste mensen die het syndroom van Gilbert hebben geen of nauwelijks klachten.
• Geelzucht: meestal treedt kort na de geboorte geelzucht op (gele huid, geelverkleuring van het oogwit). Dit verdwijnt meestal na enige tijd.
• Sommige mensen hebben af en toe buikpijn of klagen over vermoeidheid.
• Het bilirubinegehalte kan plots stijgen en de geelzucht kan opnieuw optreden, met bijhorende klachten van vermoeidheid en buikpijn, wanneer men te weinig gegeten of gedronken heeft (bijvoorbeeld na een vastenperiode), bij hevige inspanningen, wanneer men ziek is, bij een sterk verminderde weerstand en bij overmatig alcoholgebruik.
Het syndroom van Gilbert kan vastgesteld worden door middel van bloedonderzoek naar de leverfuncties. Bij het syndroom van Gilbert is alleen het bilirubinegehalte verhoogd. De andere leverfuncties zijn normaal.
De diagnose kan ook gesteld worden door middel van DNA-onderzoek.
Behandeling van het syndroom van Gilbert
Een behandeling is meestal niet nodig: een stijging van het bilirubine is niet gevaarlijk.
• Een speciaal dieet is niet nodig. Wel is het belangrijk om regelmatig te eten en te bewegen en voldoende te slapen.
• Overmatig gebruik van alcohol kan een negatieve invloed hebben.
• Indien nodig kan uw arts een geneesmiddel (fenobarbital) voorschrijven dat zorgt voor een daling van de bilirubineconcentratie in het bloed.
Syndroom van Crigler Najjar
Net als het syndroom van Gilbert is het syndroom van Crigler Najjar een erfelijke leveraandoening. Maar in tegenstelling tot het syndroom van Gilbert is het geen onschuldige aandoening. Het is gelukkig een uiterst zeldzame aandoening.
Hoe ontstaat het syndroom van Crigler Najjar?
Net zoals bij de ziekte van Gilbert breekt de lever onvoldoende bilirubine af door een tekort aan een bepaald enzyme. Bilirubine is een afvalstof die ontstaat bij de afbraak van rode bloedcellen.
Rode bloedcellen hebben een gemiddelde levensduur van ca. 6 weken. Nadien worden ze afgebroken en uit het lichaam verwijderd. Bij die afbraak wordt de rode kleurstof in het bloed, het hemoglobine, afgebroken tot bilirubine, een gele kleurstof die het lichaam verlaat via de urine en de ontlasting.
Bij het syndroom van Crigler Najjar ontbreekt een bepaald eiwit (het UDP-glucuronosyltransferase enzyme) dat de lever nodig heeft om het bilirubine te kunnen afbreken. Daardoor hoopt dat bilirubine zich op in het bloed.
In vergelijkbaar met het goedaardige syndroom van Gilbert gaat het syndroom van Crigler Najjar gepaard met veel hogere bilirubine waarden in het bloed. Als de aandoening niet tijdig wordt behandeld, kunnen hersenbeschadigingen ontstaan.
Erfelijke ziekte
Het syndroom van Crigler Najjar is een erfelijke stofwisselingsziekte die autosomaal recessief wordt geërfd.
• Autosomaal betekent dat het een niet-geslachtsgebonden aandoening is: het afwijkende gen bevindt zich niet op het 23ste chromosomenpaar dat het geslacht bepaalt. Zowel mannen als vrouwen kunnen het syndroom dus doorgeven en jongens en meisjes hebben evenveel kans om het van hun ouders te erven.
• Recessief betekent dat twee afwijkende genen nodig zijn om de ziekte te krijgen: een kind kan alleen met het syndroom geboren worden als beide ouders er zelf aan lijden of drager zijn. Dragers hebben zelf de ziekte niet en dus ook geen klachten.
Als beide ouders dragers zijn, is kans dat ook het kind de ziekte krijgt 25% en 50% dat het drager wordt (zonder de ziekte te hebben). De kans dat het kind de ziekte niet erft is eveneens 25%.
Symptomen en gevolgen van het syndroom van Crigler Najjar
Er zijn twee typen Crigler-Najjar. Type 1 is de ernstigste vorm en type 2 is minder ernstig. Bij type 2 hebben de patiënten soms nog wat restactiviteit van het gebrekkige enzym.
Patiëntjes met Crigler-Najjar krijgen meestal kort na de geboorte de eerste symptomen van geelzucht (gele huid, geel oogwit). Zonder behandeling kan de ophoping van bilirubine ernstige hersenbeschadigingen veroorzaken en er uiteindelijk toe leiden dat patiëntjes jong overlijden.
Bij infecties, bloeduitstortingen en botbreuken komt er extra bilirubine vrij, wat tot zeer ernstige opstoten van de ziekte en zelfs acuut levensgevaar kan leiden.
De diagnose kan worden gesteld met bloedonderzoek. Ook kan een stukje weggenomen lever (biopt) onderzocht worden, om de diagnose te bevestigen
Behandeling van het syndroom van Crigler Najjar
De ziekte is niet te genezen. Men kan alleen de symptomen trachten te bestrijden en de complicaties proberen te voorkomen.
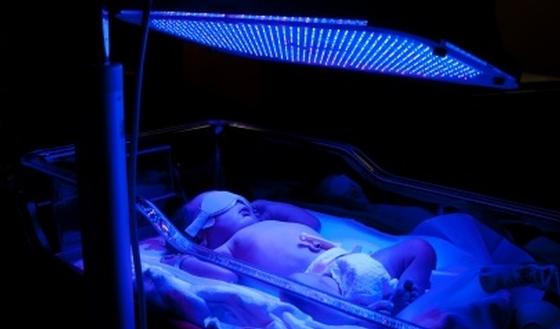
• Lichttherapie
Bilirubine wordt onder invloed van licht afgebroken. De patiëntjes moeten elke nacht onder fel blauwe lampen liggen, zoals onder een zonnebank.
Dit is niet alleen een erg belastende behandeling, men weet ook niet hoelang ze effect heeft. Vaak stijgt de concentratie van bilirubine in het bloed toch langzaam vanaf de puberteit.
Bovendien heeft deze intensieve lichtbehandeling heel wat neveneffecten: de huid wordt dunner en is erg kwetsbaar voor schrammen, wondjes en bloeduitstortingen. Wonden helen slecht en laten vaak littekens achter.
• Geneesmiddelen
Bij patiënten met type 1 (bij wie het enzym nog een beetje werkt) kan phenobarbital helpen. Dat zorgt voor een afname van bilirubine in het bloed
• Bloedtransfusie
Pasgeborenen, die hele hoge bilirubinewaarden hebben, krijgen soms een bloedtransfusie.
• Levertransplantatie
Soms zal een levertransplantatie worden uitgevoerd. Dit is echter niet zonder risico. Bovendien moet het kind levenslang geneesmiddelen nemen tegen het afstoten van de lever.
Bronnen
www.mlds.nl/ziekten
www.mlds.nl/ziekten/crigler-najjar-ziekte
http://www.stofwisselingsziekten.nl
www.najjar.nl
w.nhs.uk













